





Sickle cell disease:
Sickle cell disease(SCD) and its variants are genetic disorders resulting from the presence of a mutated form of hemoglobin, hemoglobin S (HbS) (see the image below). The most common form of SCD found in North America is homozygous HbS disease (HbSS), an autosomal recessive disorder first described by Herrick in 1910. SCD causes significant morbidity and mortality, particularly in people of African and Mediterranean ancestry (see Pathophysiology). Morbidity, frequency of crisis, degree of anemia, and the organ systems involved vary considerably from individual to individual.
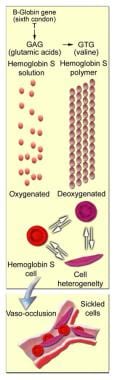 Molecular and cellular changes of hemoglobin S.
Molecular and cellular changes of hemoglobin S.Signs and symptoms
Sickle cell disease (SCD) usually manifests early in childhood. Complaints may include the following:
- Acute and chronic pain in any body part: The most common clinical manifestation of SCD is vaso-occlusive crisis; pain crises are the most distinguishing clinical feature of SCD
- Bone pain: The long bones of the extremities are often involved, often due to bone marrow infarction
- Anemia: Universally present, chronic, and hemolytic in nature
- Aplastic crisis: Serious complication due to infection with B19V
- Splenic sequestration: Characterized by the onset of life-threatening anemia with rapid enlargement of the spleen and high reticulocyte count
- Infection: Organisms that pose the greatest danger include encapsulated respiratory bacteria, particularly Streptococcus pneumonia; adult infections are predominately with gram-negative organisms, especially Salmonella
- Growth retardation, delayed sexual maturation, being underweight
- Hand-foot syndrome: This is a dactylitis presenting as bilateral painful and swollen hands and/or feet in children
- Acute chest syndrome: Young children present with chest pain, fever, cough, tachypnea, leukocytosis, and pulmonary infiltrates in the upper lobes; adults are usually afebrile, dyspneic with severe chest pain, with multilobar/lower lobe disease
- Pulmonary hypertension: Increasingly recognized as a serious complication of SCD
- Avascular necrosis of the femoral or humeral head: This is due to vascular occlusion
- CNS involvement: Most severe manifestation is stroke
- Ophthalmologic involvement: Ptosis, retinal vascular changes, proliferative retinitis
- Cardiac involvement: Dilation of both ventricles and the left atrium
- GI involvement: Cholelithiasis is common in children; liver may become involved
- GU involvement: Kidneys lose concentrating capacity; priapism is a well-recognized complication of SCD
- Dermatologic involvement: Leg ulcers are a chronic painful problem
Triggers of vaso-occlusive crisis include the following:
- Hypoxemia: May be due to acute chest syndrome or respiratory complications
- Dehydration: Acidosis results in a shift of the oxygen dissociation curve
- Changes in body temperature (eg, an increase due to fever or a decrease due to environmental temperature change)
See Clinical Presentation for more detail.
Diagnosis
SCD is suggested by the typical clinical picture of chronic hemolytic anemia and vaso-occlusive crisis. Electrophoresis confirms the diagnosis with the presence of homozygous HbS and can also document other hemoglobinopathies (eg, HbSC, HbS-beta+ thalassemia).
Laboratory tests used in patients with SCD include the following:
- Mandatory screening for HbS at birth in the United States; prenatal testing can be obtained via chorionic villus sampling
- Hemoglobin electrophoresis
- CBC count with differential and reticulocyte count
- Serum electrolytes
- Hemoglobin solubility testing
- Peripheral blood smear
- Pulmonary function tests (transcutaneous O 2 saturation)
- Renal function (creatine, BUN, urinalysis)
- Hepatobiliary function tests, (ALT, fractionated bilirubin)
- CSF examination: Consider LP in febrile children who appear toxic and in those with neurologic findings (eg, neck stiffness, + Brudzinski/Kernig signs, focal deficits); consider CT scanning before performing LP
- Blood cultures
- ABGs
- Secretory phospholipase A2 (sPLA2)
In one study of 38 asymptomatic children with SCD, investigators found that hypertension and abnormal blood pressure patterns were prevalent in children with SCD.[1] They suggested using 24-hour ambulatory BP monitoring (ABPM) to identify these conditions in young patients.[1]
In the study, 17 patients (43.6%) had ambulatory hypertension, whereas 4 (10.3%) had hypertension on the basis of their clinic blood pressure. Twenty-three patients (59%) had impaired systolic blood pressure dipping, 7 (18%) had impaired diastolic blood pressure dipping, and 5 (13%) had reversed dipping.[1]
Imaging studies
Imaging studies that aid in the diagnosis of sickle cell anemia in patients in whom the disease is suggested clinically include the following:
- Radiography: Chest x-rays should be performed in patients with respiratory symptoms
- MRI: Useful for early detection of bone marrow changes due to acute and chronic bone marrow infarction, marrow hyperplasia, osteomyelitis, and osteonecrosis
- CT scanning: May demonstrate subtle regions of osteonecrosis not apparent on plain radiographs in patients who are unable to have an MRI [2] and to exclude renal medullary carcinoma in patients presenting with hematuria
- Nuclear medicine scanning: 99m Tc bone scanning detects early stages of osteonecrosis; 111 In WBC scanning is used for diagnosing osteomyelitis
- Transcranial Doppler ultrasonography: Can identify children with SCD at high risk for stroke
- Abdominal ultrasonography: Can be used to rule out cholecystitis, cholelithiasis, or an ectopic pregnancy and to measure spleen and liver size
- Echocardiography: Identifies patients with pulmonary hypertension
- Transcranial near-infrared spectroscopy or cerebral oximetry: Can be used as a screening tool for low cerebral venous oxygen saturation in children with SCD
See Workup for more detail.
Management
The goals of treatment in SCD are symptom control and management of disease complications. Treatment strategies include the following 7 goals:
- Management of vaso-occlusive crisis
- Management of chronic pain syndromes
- Management of chronic hemolytic anemia
- Prevention and treatment of infections
- Management of the complications and the various organ damage syndromes associated with the disease
- Prevention of stroke
- Detection and treatment of pulmonary hypertension
Pharmacotherapy
SCD may be treated with the following medications:
- Antimetabolites: Hydroxyurea
- Opioid analgesics (eg, oxycodone/ASA, methadone, morphine sulfate, oxycodone/APAP, fentanyl, nalbuphine, codeine, APAP/codeine)
- Nonsteroidal analgesics (eg, ketorolac, ASA, APAP, ibuprofen)
- Antibiotics (eg, cefuroxime, amoxicillin/clavulanate, penicillin VK, ceftriaxone, azithromycin, cefaclor)
- Vaccines (eg, PCV7, PPV23, meningococcal, influenza, recommended scheduled childhood/adult vaccinations)
- Vitamins (eg, folic acid)
Nonpharmacologic therapy
Other approaches to managing SCD include the following:
- Stem cell transplantation: Can be curative
- Transfusions: For sudden, severe anemia due to acute splenic sequestration, parvovirus B19 infection, or hyperhemolytic crises
- Physical therapy
- Heat and cold application
- Acupuncture and acupressure
- TENS
Combination pharmacotherapy and nonpharmacotherapy
- Vigorous hydration (plus analgesics): For vaso-occlusive crisis
- Oxygen, antibiotics, analgesics, incentive spirometry, simple transfusion, and bronchodilators: For treatment of acute chest syndrome
0 التعليقات:
Post a Comment