





Background
Congenital diaphragmatic hernia (CDH) affects approximately 1 in 2000 newborns. Although great strides have been made in the management of CDH, significant morbidity and mortality persist.
The current mainstay of treatment of CDH involves stabilization and respiratory support at birth, followed by closure of the diaphragmatic defect, which returns the abdominal organs to the abdominal cavity and makes room in the chest for the hypoplastic lung to grow. While some specialized centers report a survival rate of close to 90% with this technique, pooled results from over 50 centers worldwide indicate the overall survival rate to be 69% with this standard postnatal therapy (CDH study group , October 2010).
Specific morbidities in survivors include neurodevelopmental, nutritional, sensorineural hearing, and pulmonary function deficiencies, all of which are most likely attributable to the severity of lung hypoplasia and pulmonary hypertension that accompany CDH. The severe consequences of this congenital anomaly have led others to pursue methods of correcting CDH or its main consequence, lung hypoplasia, prior to birth, allowing for more normal postnatal function.
Fetal surgery for CDH was originally proposed to involve open fetal surgery and repair of the diaphragmatic defect, similar to postnatal surgical care, in mid gestation. Indeed, today, there are a handful of survivors of open fetal CDH repair.[3]
However, as advances were made in neonatal care, the postnatal survival rate among patients undergoing standard care (without fetal intervention) improved. In addition, pioneers in this endeavor found that fetuses who could benefit most from fetal repair were also those who were most likely to die of the procedure.
The main challenge for these fetuses was that the liver was herniated into the chest, and, when the liver was reduced back into the abdomen during open fetal repair, the blood flow through the umbilical vein and inferior vena cava became occluded, resulting in death of the fetus.[4] Fetuses without liver herniation typically had sufficient lung development to ensure a relatively good outcome with standard postnatal care.
As investigators struggled with this challenge, two main phenomena resulted in the evolution of the current paradigm of fetal intervention for CDH: (1) the advent of minimally invasive surgical procedures and (2) recognition of a condition called congenital high airway obstruction syndrome (CHAOS), in which fetuses whose airways were occluded either by a tumor or an atresia developed oversized lungs.
In the 1990s, the development and implementation of minimally invasive surgical techniques advanced at a rapid pace. The manufacture of small telescopes and various new surgical instruments allowed for further innovations. Simultaneously, irritation of the uterus and the threat of preterm labor were identified as major hurdles in fetal interventions.
Reasoning that minimal manipulation of the uterus would help reduce the risk of preterm labor, the technique of fetoscopic tracheal occlusion to promote lung growth was conceived. Initial studies demonstrated that manipulation of mechanical forces involved in lung development, namely the lung fluid production that distends the airways, could be applicable in the treatment of CDH.[5, 6, 7]
Additional laboratory investigations, mainly using the fetal lamb model of CDH, confirmed the feasibility and effectiveness of the tracheal occlusion procedure, leading to the first human interventions.[8, 9, 10, 11, 12, 13] Initially, a surgical clip was placed on the trachea through a neck incision. Although this technique effectively occluded the trachea, scarring and the development of tracheal stenosis were identified as significant adverse effects. Ultimately, fetal bronchoscopy with tracheal balloon occlusion emerged as an appropriate solution. See image below.
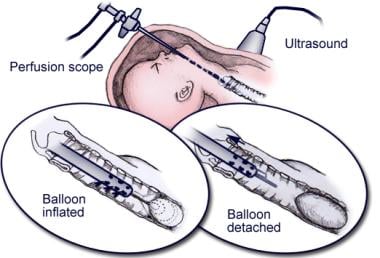 Schematic of fetal tracheal balloon occlusion, using a catheter-based detachable balloon that is placed through the side channel of the fetoscope.
Schematic of fetal tracheal balloon occlusion, using a catheter-based detachable balloon that is placed through the side channel of the fetoscope.
Once the technique was refined, a National Institutes of Health (NIH)–sponsored randomized controlled trial of fetal tracheal occlusion versus standard postnatal care for severe CDH was conducted.[14] Although this trial did not demonstrate a survival advantage with tracheal occlusion, it was notable that the group who underwent tracheal occlusion was born significantly earlier than the control group. In addition, despite this prematurity, lung function in infancy was demonstrated to be possibly slightly improved compared to the control group.[15]
These findings have led to worldwide enthusiasm for the fetal balloon tracheal occlusion technique.[16, 17, 18] It is important to note, however, that this enthusiasm partly results from an inability to otherwise improve the overall survival for CDH over the past two decades, as well as the significant comorbidities among survivors of severe CDH.[2] Strictly speaking, fetal tracheal occlusion for CDH remains an investigational therapy for which the long-term benefits have yet to be proven.
Indications
Identification of fetuses with congenital diaphragmatic hernia (CDH) who may benefit from fetal tracheal occlusion has been a major challenge. Once the diagnosis of CDH is confirmed by an experienced radiologist using either ultrasonography or MRI, a careful evaluation must be conducted for any other major anomalies, such as cardiac defects or chromosomal abnormalities. In addition, any maternal operative risk factors, such as any significant underlying medical conditions, must be considered.
If all of these contraindications are excluded, further consideration is given to fetal intervention. The main prenatal determinants concerning the severity of isolated CDH used today are the LHR and the presence or absence of liver herniation.
The LHR is a measurement that can be made with ultrasonography or MRI and quantifies the relative size of the lungs.[19, 20] An LHR of 1.0 or less signifies severe lung hypoplasia due to the CDH.
Liver herniation is determined using the same imaging modalities; at UCSF, the presence of liver herniation is required for consideration of the fetal tracheal occlusion procedure. Recent evidence indicates that the amount of liver herniation into the chest may be a better predictor of disease severity than simple presence or absence of liver herniation.[21]
A complete listing of inclusion and exclusion criteria for fetal intervention for CDH is outlined below and in Contraindications.
The following are inclusion criteria for fetal surgery for CDH:
- Confirmed diagnosis of CDH
- Normal fetal echocardiography result
- Normal karyotype
- Liver is herniated into the left hemithorax
- Lung-to-head ratio (LHR) of 1.0 or less, calculated between 24 and 26 weeks' gestation
- Fetus is 26-28 weeks' gestation
- Singleton pregnancy
- Maternal age of 18 years or older
- Meets psychosocial criteria as determined by social worker screening
- Preauthorization from a third-party payor for fetal intervention OR the ability to self-pay for study treatment
Contraindications
The following are exclusion criteria for fetal surgery for CDH:
- Failure to meet all inclusion criteria
- Other congenital anomalies detected on ultrasonography
- Contraindication to abdominal surgery or general anesthesia
- Preterm labor, preeclampsia, or uterine anomaly (eg, large fibroid tumor)
- Family unable or refuses to stay near medical center for the duration of the tracheal occlusion period and for the duration of the pregnancy as medically necessary
Outcomes
Since the original randomized controlled trial of fetal endoscopic tracheal occlusion performed at UCSF,[14] other groups have demonstrated an early survival benefit.[25, 26] These studies are limited by the complex nature of the postnatal management of patients with CDH and numerous variables that are difficult to control, but, overall, the therapy remains very promising for severely affected fetuses. Long-term functional outcomes following fetal tracheal occlusion for CDH have yet to be reported.
The most significant potential complications, specifically quantified in the aforementioned 3 studies, include maternal death (0%), fetal demise (0%-2%), preterm premature rupture of membranes (35%-100%), and preterm delivery (50%-73%), with an average gestational age at delivery of 31 weeks in the Harrison series and 35 weeks in the more recent series. Tracheomegaly and tracheobronchomegaly have also been reported with a fairly high incidence.[27, 28]For most of these patients, there are minimal symptoms, such as a barking cough, although patients who require further airway procedures have been reported
Fetal Surgery for Congenital Diaphragmatic Hernia Periprocedural Care
Patient Education & Consent
The risks, benefits, and potential complications are thoroughly reviewed with the patient.
Pre-Procedure Planning
Appropriate consultations are made with the pediatric surgeon, perinatologist, anesthesiologist, and social worker.
Patient Preparation
The patient is admitted to the obstetrical unit, and a baseline physical assessment is performed, including fetal heart rate and uterine contraction monitoring. The latest prenatal sonogram is reviewed and indications for surgery are confirmed.
Preoperative laboratory tests include a complete blood cell count, blood type and screen, and urinalysis. Activity is ad lib and bedrest exercises are reviewed.
The patient is kept nil per os for 8 hours before surgery.
Immediately before surgery, indomethacin for tocolysis may be administered, and perioperative antibiotics are given.
Anesthesia
Depending on the surgeon's choice, anesthesia can range from local to locoregional to general.
Monitoring & Follow-up
After the procedure, the patient is readmitted to the obstetrical unit for monitoring.
Daily ultrasonography and fetal echocardiography (if indicated) are performed. After the procedure on the day of surgery, the patient's diet is advanced as tolerated, and she remains in bed with lateral positioning. Owing to potential pulmonary edema, fluids are restricted to 3 L/d if the patient is treated with magnesium sulfate. Incentive spirometry and continuous pulse oximetry are instituted.
Fetal heart rate and uterine contractions are monitored until discontinued by the fetal treatment team. Tocolysis is achieved with indomethacin, magnesium sulfate, or nifedipine, as dictated by the fetal treatment team. On postoperative day number 1, the patient is allow to ambulate and undergoes a complete blood cell count.
The patient is discharged when she is ambulatory, is tolerant of a diet, is spontaneously voiding, has good pain control with oral medications, is afebrile with normal vital signs and reassuring physical examination findings, and has a normal fetal heart rate and minimal uterine contractions. Oral tocolysis is continued as needed.
Reversal Procedure
Since the development of the fetal tracheal occlusion technique, reversal of the occlusion has been recognized as an important part of the therapy, both to avoid the necessity of an ex utero intrapartum treatment (EXIT) procedure[22] for delivery and to allow the lungs to recover from the distention forces that result in lung growth.[23]
Reversal is achieved by repeat fetoscopy and balloon removal 4-6 weeks after the initial procedure. In the future, more sophisticated tracheal occlusion devices may allow for more physiologic lung growth and development.[24]
Fetal Surgery for Congenital Diaphragmatic Hernia Technique
Fetal Surgery for Congenital Diaphragmatic Hernia
After appropriate counseling has been completed, and risks, benefits, and potential complications have been thoroughly reviewed and the family wishes to proceed, preparations are made for the fetal bronchoscopy and tracheal occlusion procedure.
The technique was originally described by Harrison et al[13] and more recently in detail by Deprest et al.[31]Briefly, after appropriate anesthesia is achieved, the uterine and fetal anatomy are carefully surveyed with ultrasonography.
The placenta is avoided, and a laparoscopic trochar (usually a disposable central venous introducer sheath) is inserted using the Seldinger technique.
The fetoscope is introduced and maneuvered into the oropharynx, through the glottis, and into the trachea. The detachable balloon catheter is passed through the side-channel of the fetoscope, the balloon is inflated and detached, and then the fetoscope is withdrawn.
Appropriate positioning of the balloon in the trachea is confirmed by ultrasound. See the multimedia files below.
This feature requires the newest version of Flash. You can download it here.
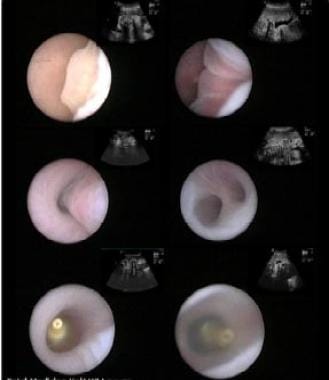 Fetoscopic images of landmarks during percutaneous fetal tracheal balloon occlusion. From top left to right bottom: epiglottis, vocal cords, trachea, carina, inflated and detached balloon, vocal cords about to close over the balloon. Image courtesy of Division of Pediatric Surgery, University of California, San Francisco, School of Medicine.
Fetoscopic images of landmarks during percutaneous fetal tracheal balloon occlusion. From top left to right bottom: epiglottis, vocal cords, trachea, carina, inflated and detached balloon, vocal cords about to close over the balloon. Image courtesy of Division of Pediatric Surgery, University of California, San Francisco, School of Medicine.
0 التعليقات:
Post a Comment